
More Information
Submitted: July 31, 2023 | Approved: September 14, 2023 | Published: September 15, 2023
How to cite this article: Kaler AK, Bora NS, Kavyashree P, Nikam A, Rane S, et al. A Case of X-Linked Hypophosphatemia: Exploring the Burden in a Single Family and the Significance of a Multidisciplinary Approach. Arch Case Rep. 2023; 7: 042-045.
DOI: 10.29328/journal.acr.1001076
Copyright License: © 2023 Kaler AK, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Hypophosphatemia; PHEX; FGF23; Rickets

A Case of X-Linked Hypophosphatemia: Exploring the Burden in a Single Family and the Significance of a Multidisciplinary Approach
Amrit Kaur Kaler1, Nandini Shyamali Bora2*, Kavyashree P3, Ankita Nikam3, Samrudhi Rane4, Yash Tiwarekar4, Shweta Limaye5 and Archana Juneja6
1Consultant, Department of Molecular Pathology and Genomics, Kokilaben Dhirubhai
Ambani Hospital, Mumbai, Maharashtra, India
2Genetic Counsellor, Department of Molecular Pathology and Genomics, Kokilaben
Dhirubhai Ambani Hospital, Mumbai, Maharashtra, India
3Technical Officer, Department of Molecular Pathology and Genomics, Kokilaben
Dhirubhai Ambani Hospital, Mumbai, Maharashtra, India
4Junior Technical Officer, Department of Molecular Pathology and Genomics,
Kokilaben Dhirubhai Ambani Hospital, Mumbai, Maharashtra, India
5Senior Technical Officer, Department of Molecular Pathology and Genomics,
Kokilaben Dhirubhai Ambani Hospital, Mumbai, Maharashtra, India
6Consultant, Department of Diabetes & Bariatric Surgery, Endocrinology & Diabetes,
Kokilaben Dhirubhai Ambani Hospital, Mumbai, Maharashtra, India
*Address for Correspondence: Nandini Shyamali Bora, M.Sc, Genetic Counsellor, Department of Molecular Pathology and Genomics, Dahanukadwadi, Kandiwali West 400067, Mumbai, Maharashtra, India, Email: [email protected]
A 46-year-old lady was diagnosed clinically with X-linked hypophosphatemia (XLH) with a rare pathogenic variant detected using exome sequencing. Phosphate-regulating endopeptidase homologous X linked (PHEX) is normally expressed in osteoblasts and osteocytes, and senses phosphate regulation. More than 1000 PHEX variants have been detected to date, which are caused by missense, nonsense, and frameshift mutations in addition to splice variants and copy number changes. The aberration in the PHEX gene leads to the upregulation of fibroblastic growth factor 23 (FGF23), which leads to defects in phosphate metabolism. This results in impaired bone growth and mineralization, short and disproportionate stature, leg bowing, musculoskeletal pain, spontaneous dental abscesses, rickets, and osteomalacia in XLH patients. The spectrum of manifestations differs between pediatric and adult patients. In our case study, two of the patient’s children started showing symptoms at a younger age, unlike their mother. Timely diagnosis and the start of treatment would help in their better management and improved quality of life.
XLH: X-Linked Hypophosphatemia; PHEX: Phosphate Regulating Endopeptidase Homologous X linked; FGF23: Fibroblast-Growth-Factor 23; HGMD: Human Gene Mutation Database; PTH: Parathyroid Hormone; TmP: Tubular Maximum Reabsorption of Phosphate to; GFR: Glomerular Filtration Rate; FDG: Fluorodeoxy Glucose; PET-CT: Positron Emission Tomography and Computed Tomography; OMIM: Online Mendelian Inheritance in Man; LSDB: Locus-Specific Data Bases; LOVD: Leiden Open Variation Database; ncRNA: non-coding RNA; UTR: Untranslated Region
X-Linked Hypophosphatemia (XLH) is the most common cause of inherited rickets and is transmitted in an X X-linked dominant pattern caused by a defect in the PHEX gene (phosphate regulating endopeptidase homologous X-linked). PHEX gene is located on chromosome Xp22.1 and shows complete penetrance in both males and females but no male-to-male transmission [1]. Genetic variations in the PHEX gene lead to increased circulating fibroblast-growth-factor 23 (FGF23) levels, which causes decreased renal absorption and deranged vitamin D3 metabolism in the body, leading to defective mineralization. Excessive FGF23 activity leads to increased phosphate excretion in the kidneys, mediated by the downregulation of renal tubular sodium phosphate co-transporters (2a/2c), and reduced phosphate absorption in the intestines due to impaired vitamin D activation, leading to fractures and inadequate mineralization (Figure 1) [2,3]. More than 1000 PHEX variants have been reported and are available on the Human gene mutation database (HGMD, https://www.hgmd.cf.ac.uk/ac/all.php) [4]. The clinical presentation of XLH is variable based on the pathogenicity of the variant. The typical symptoms might include isolated hypophosphatemia, rickets, osteomalacia, short stature, enthesopathy and hearing loss, severe leg bowing leading to bone pain, and reduction of daily activities [5]. The genotype-phenotype correlation will help us understand the pathogenesis of the disease and hence plan the treatment of these patients.
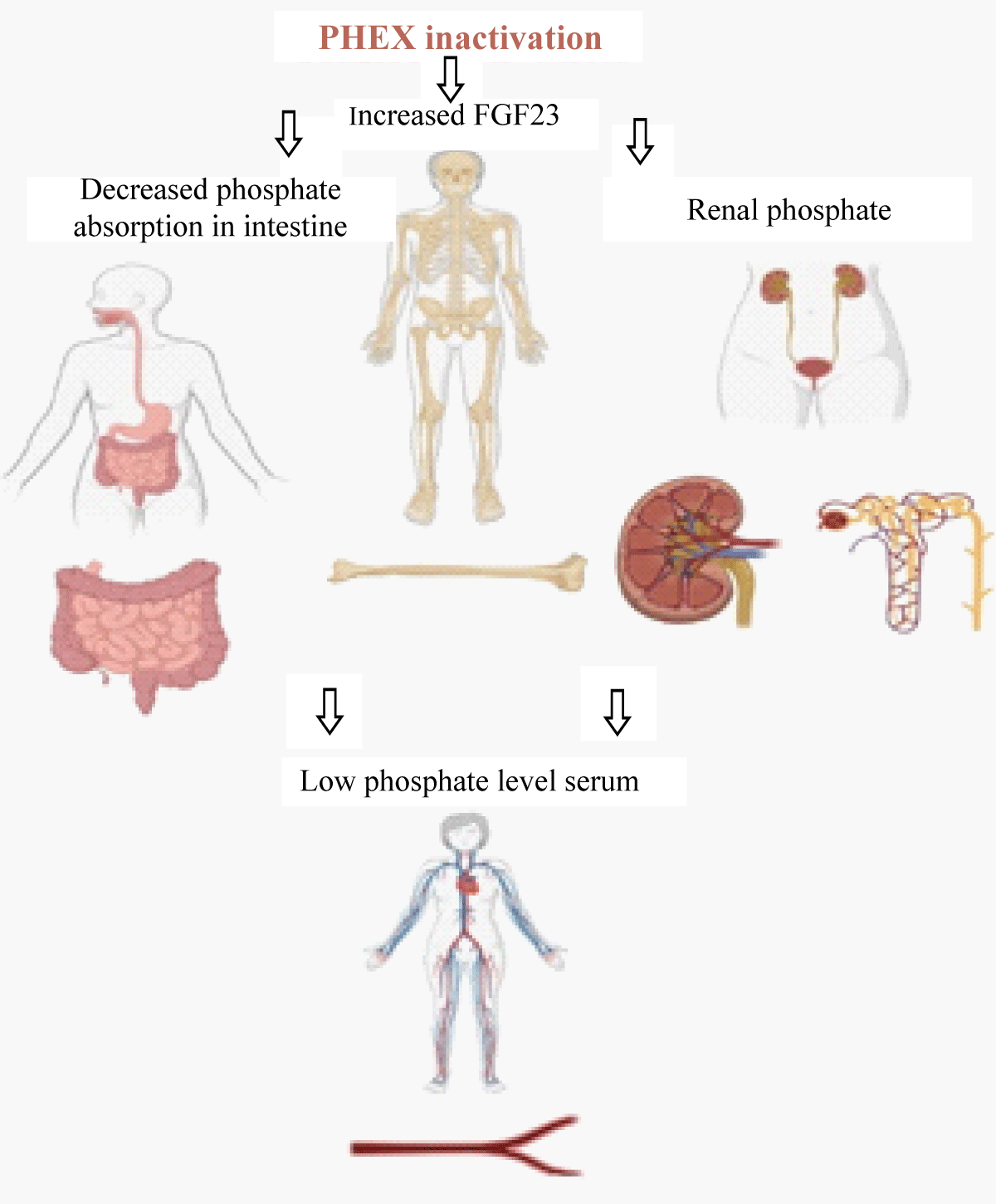
Figure 1: Schematic diagram showing the affects of PHEX gene inactivation.
A 46-year-old female born to non-consanguineous parents, presented with clinical indications of hypophosphatemia, hyperthyroidism, severe osteomalacia, osteoporosis, joint pain, and inability to walk for the last 8 years. She had gait difficulty, difficulty in climbing stairs, getting up from squatting or sitting position, lower limb varus deformities starting from her early thirties and is now non-ambulant. Laboratory investigations showed low levels of blood creatinine (0.30 mg/dl), low phosphorous (1.90 mg/dl), high serum parathyroid hormone (PTH) (108 pg/ml), and near normal D3 levels. She is a known case of hyperparathyroidism and has a past history of multiple fractures with malunion. MRI spine showed diffuse demineralization (osteoporosis) with multiple pseudo-fractures and sustained old fractures involving ribs, vertebrae, pelvic bones, proximal femoribilateral iliac bones, bilateral intertrochanteric region, bilateral pubic rami, suggestive of osteomalacia. Additional degenerative changes in the form of a few multilevel anterior marginal osteophytes and disc desiccation posterior disc bulges at C3-C4, C4-C5, C5-C6, L1-L2, L2-L3, and L3-L4 were noted. The patient was on oral calcium and cholecalciferol supplements daily with cholecalciferol capsules (60000 IU) weekly for 6-8 weeks intermittently over the last 5 years.
The ratio of tubular maximum reabsorption of Phosphate (TmP) to Glomerular Filtration Rate (GFR) on urine samples used to evaluate renal phosphate levels was found to be low, indicating renal phosphate loss. A Fluorodeoxyglucose (FDG) PET-CT scan was done which showed an ill-defined, hypoenhancing, non-avid area in the pancreatic head with a mildly prominent pancreatic duct. 68 Ga-DOTA-NOC scan was used to rule out Tumor-induced osteomalacia associated with neuroendocrine tumors.
Family history: The patient has a twenty-year-old son who started showing symptoms of walking difficulty in the last five years and now walks with support; a normal healthy daughter of 18 years age and a younger daughter aged fourteen years age who underwent correction surgery for bowed legs two years back. The patient was suggested to undergo genetic testing and counseled regarding the test and the implications of the test results for her and her family members. The patient’s parents were normal with no symptoms.
Exome sequencing of the patient revealed a pathogenic heterozygous variant in the PHEX gene (c.1645+1G >A) which is associated with Hypophosphatemic Rickets (OMIM #307800). The variant is a 5’ splice site variation in intron 15 of the PHEX gene, that affects the invariant GT donor splice site of exon 15 and is expected to disrupt RNA splicing. The observed variation has previously been reported in patients affected with hypophosphatemia rickets and has been classified as pathogenic in the ClinVar database (RCV000505469.4). This variant has not been reported in the 1000 genomes and gnomAD databases. The in-silico prediction of the variant is damaged by the MutationTaster2 tool. Since the disorder is transmitted in an X-linked dominant manner, for an affected female there is a 50% chance in each pregnancy to pass this variant to her children. A male affected with this condition will pass this variant to all of his daughters but not his sons. Due to intrafamilial variation, the severity of the disorder cannot be predicted in the affected individuals carrying the variant. Sanger validation of the detected variant and segregation analysis in parents was recommended. The patient was referred to the endocrinologist and orthopaedician for further follow-up. Targeted testing of the detected variant in affected and unaffected children was recommended. If the PHEX pathogenic variant is detected in an affected family member, they were encouraged to follow up for pre-conceptional/prenatal genetic counseling.
The patient was treated with phosphate granules and calcitriol supplementation by the endocrinologist. Phosphate sachets (3.2 gm/sachet) were added which is equivalent to 500 mg phosphate, started once daily and titrated up to 2 sachets per day. The patient is also receiving daily doses of 0.25 mg of calcitriol once. Current levels of serum phosphorus and calcium are maintained between 2.2 - 2.4 mg/dl and 8 - 8.5 mg/dl on treatment. The patient was advised for three monthly follow-ups with serum Calcium / Phosphate and 24-hour urine calcium and creatinine test.
X-linked hypophosphatemia (XLH) is a genetic, phosphate-wasting progressive disease with an estimated incidence of 1:20,000 - 25,000 individuals. The genetic association was first reported 25 years back and till now more than 1000 variants have been reported in the literature [4]. Pathogenic variations like missense, nonsense, frameshift, copy number, and splice site variants have been identified throughout the entire length of the gene. This leads to a loss of function in the phosphate-regulating endopeptidase homolog X-linked (PHEX) gene, which leads to truncating and non-truncating proteins. PHEX gene is known to regulate the zinc metallopeptidases involved in bone renal diseases, arthritis, inflammatory disorders, cardiovascular diseases, cancer, etc. PHEX gene codes for a transmembrane endopeptidase and is primarily expressed by osteoblasts, osteocytes, and odontoblasts in bones and teeth. As a result of pathogenic mutations in the PHEX gene, there is an increase in the serum level of Fibroblast growth factor 23 (FGF23), which is involved in phosphate homeostasis in the body. However, the cause of this increase is still unclear [6].
High FGF23 internalizes the renal sodium-phosphate transporters which results in hypophosphatemia and phosphate wasting; downregulates the renal 1 alpha-hydroxylase enzyme and upregulates the 24-hydroxylase enzyme thus impairing the synthesis of 1, 25(OH)2 vitamin D and increases its degradation [4]. These defects in phosphate metabolism thus result in fractures and inadequate bone mineralization, short stature, enthesopathy, severe leg bowing, spontaneous dental abscesses, rickets, and osteomalacia. The spectrum of manifestations has been seen to differ among different patients based on different pathogenic variants [7,8].
Although XLH has been known to be associated with the PHEX gene, the variant detected in the present case is the first to be reported from India. This variant (NM_000444.6 (PHEX): c.1645+1G>A) is a single nucleotide 5’splice site variant mutation which is a heterozygous variant in intron15 of the PHEX gene. The possible hypothesis for pathogenicity is that it could generate partial splicing errors which might result in the inactivation of the PHEX gene and hence explain the genotype-phenotype correlation [1].
Clinvar has given 4 submissions for the variant which have been reported as Pathogenic. This variant is not reported in the gnomAD database and 1000 genomes. In the study by Zhang et al., 2019 only 3.23% of their study cohort of 261 patients showed the mutation [8]. Popowska, et al. 2000 in their study of 29 Polish patients with XLH, reported this variant in three female patients [9]. Fahiminiya S et al.,2014 studied 17 consanguineous families in Qatar with diverse genetic disorders and reported the variant in one member(daughter) as a de novo mutation with non-carrier parents [10].
Different databases have been created to understand the gene variants of the PHEX gene. Locus-specific databases https://grenada.lumc.nl/LSDB_list/lsdbs/ phexhas been created for the PHEX gene which gives a complete database and accurate information on unique variants based on Clinvar, Global Variome shared Leiden Open Variation Database (LOVD), PHEXdb Locus Database etc. ClinVar has reported 1316 unique PHEX variants as of March 5, 2023. Out of them, 700 are single nucleotide variants, 373 are deletions, 201 are duplications, 10 are indels and 149 are insertions. Molecular consequence wise Frameshift(213), Missense(249), Nonsense(146), Splice site(162), ncRNA(66) and UTR(57) have been reported. Global Variome shared Leiden Open Variation Database (LOVD) has reported 1042 unique PHEX gene variants as of Jan 11, 2023 (https://databases.lovd.nl/shared/genes/PHEX)
Conventional treatment for patients with XLH consists of active vitamin D analogs and oral phosphate supplements as a mainstay with calcium supplementation, under regular monitoring [11]. The risk of complications arising from phosphate therapy, such as hyperparathyroidism, nephrocalcinosis, hypercalciuria as well as gastrointestinal side effects is reported in patients which might call for additional interventions. Though these therapies have helped in improving symptoms, it is seen that there is persistence of short stature, skeletal malformation, and incomplete recovery from rickets associated with XLH [12]. The patient was treated with phosphate granules(3.2 gm/sachet) and 0.25 mg of calcitriol supplementation started once daily and titrated up to 2 sachets per day. The patient was on a wheel wheelchair but now is able to stand with support. The brother of the affected patient has taken responsibility for the treatment, follow-up, and support, and has been to multiple hospitals before ending this diagnostic odyssey.
Anabolic steroids, especially Nandrolone decanoate were popular till the last century for the treatment of postmenopausal osteoporosis in view of their anabolic properties on the bone, However, it has potentially serious adverse effects on the cardiovascular system, hypertension, liver, endocrine (infertility, virilization, gynecomastia, and hypogonadism), tendon ruptures, neuropsychiatric disturbances, and the high risk for addiction and abuse. Hence this drug is no longer recommended for the treatment of metabolic bone disorders [13].
In their study, Lo, et al. described how the typical symptoms of pain, weariness, and stiffness had frequently worsened over time for many people affected with XLH and were accompanied by anxiety about the future. The majority of participants in their study also expressed concern or guilt about having children with XLH [14].
The psychological impacts of having children with XLH and the burden of passing the disease are a primary concern among many patients along with expenses associated with the treatment and management of the disease in multiple family members [15]. Genetic counseling and testing help individuals understand the disease implications and its inheritance and the patient and their family can be made aware of the available prenatal testing options in order for the future generations to make informed decisions. The psychosocial implications of having a hereditary condition are also highlighted in this case. The feeling of hopelessness was associated with the adult onset of the condition in the family member and questioning of how and why has been noted in the genetic counseling session. Taking the help of a genetic counsellor has helped the patient and her family members to understand the disorder and plan further steps for her children. Follow-up was planned for therapy and the patient has undergone physical therapy sessions which has also improved her quality of life and reduced pain symptoms.
The case reported is the first from India with the (c.1645+1G >A) splice site variant in the PHEX gene. Given the rarity of the condition, affecting one in 20,000 individuals, the significance of a multidisciplinary team composed of endocrinologists, orthopaedic surgeons, molecular pathologists, genetic counselors, and physical therapists can help with diagnosis and in designing the appropriate management of X Linked Hypophosphatemia (XLH). Familial segregation and genetic workup for her children is recommended for securing future generations and healthcare workers are responsible for making the patient aware of the different reproductive options available to them at the right time.
The authors thank the patient and her family for their cooperation and for providing consent in the case report.
Author’s contributions
AK conceptualized the design of the case report and was involved in drafting the article. NB and KP were involved in writing the article. AN, SR, YT, SL, YP, and AJ revised the article critically for important intellectual content. All the authors approve the final version to be published.
- Ichikawa S, Traxler EA, Estwick SA, Curry LR, Johnson ML, Sorenson AH. Mutational survey of the PHEX gene in patients with X-linked hypophosphatemic rickets. Bone. 2008; 43(4):663-6.
- Ruppe MD. X-Linked Hypophosphatemia. In: Adam MP, Mirzaa GM, Pagon RA. editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2023. 2012. https://www.ncbi.nlm.nih.gov/books/NBK83985/
- Sarafrazi S, Daugherty SC, Miller N, Boada P, Carpenter TO, Chunn L. Novel PHEX gene locus-specific database: Comprehensive characterization of vast number of variants associated with X-linked hypophosphatemia (XLH). Hum Mutat. 2022; 43(2):143-157.
- Beck-Nielsen SS, Brixen K, Gram J, Brusgaard K. Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and CLCN5 in patients with hypophosphatemic rickets. J Hum Genet. 2012; 57(7):453-8.
- Song HR, Park JW, Cho DY, Yang JH, Yoon HR, Jung SC. PHEX Gene Mutations and Genotype-Phenotype Analysis of Korean Patients with Hypophosphatemic Rickets. J Korean Med Sci. 2007;22(6):981-986.
- Novel PHEX Variants and Splicing Mutations in Patients with X-Linked Hypophosphatemia. Research Square Xing X, Gao J, Ma H. 2021.
- Padidela R, Nilsson O, Makitie O. The international X-linked hypophosphataemia (XLH) registry (NCT03193476): rationale for and description of an international, observational study. Orphanet J Rare Dis. 2020; 15:172.
- Zhang C, Zhao Z, Sun Y. Clinical and genetic analysis in a large Chinese cohort of patients with X-linked hypophosphatemia. Bone. 2019; 121:212-220.
- Popowska E, Pronicka E, Sułek A, Jurkiewicz D, Rowe P, Rowinska E. X-linked hypophosphatemia in Polish patients. 1. Mutations in the PHEX gene. J Appl Genet. 2000; 41(4):293-302.
- Fahiminiya S, Almuriekhi M, Nawaz Z, Staffa A, Lepage P, Ali R. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin Genet. 2014;86(2):134-41.
- Cabrera BMJ, Chacha OPR, Reza-Albarrán AA, Chacha OAK, Acero MA, Serrano RA. X-linked hypophosphatemic rickets: Case report of late diagnosis and bone pain improvement with targeted treatment. Clin Case Rep. 2022;10(8): e6217.
- Baroncelli GI, Mora S. X-Linked Hypophosphatemic Rickets: Multisystemic Disorder in Children Requiring Multidisciplinary Management. Front Endocrinol (Lausanne). 2021 Aug 6; 12:688309. doi: 10.3389/fendo.2021.688309. PMID: 34421819; PMCID: PMC8378329.
- Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Linglart A. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nature Reviews Nephrology. 2019; 15(7):435-455.
- Lo SH, Lachmann R, Williams A, Piglowska N, Lloyd AJ. Exploring the burden of X-linked hypophosphatemia: A European multi-country qualitative study. Qual Life Res.2020;29(7):1883-1893.
- Kanamalla K, Fuchs R, Herzog C, Steigbigel KD, Macica CM. An Evidence-based Physical Therapy Prescription for Adults with X-linked Hypophosphatemia. J Endocr Soc. 2022 Jun 18; 6(8): bvac094.